|
Sequence comparisons in the internet:
Dot matrices
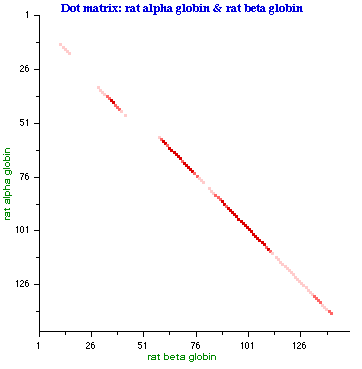 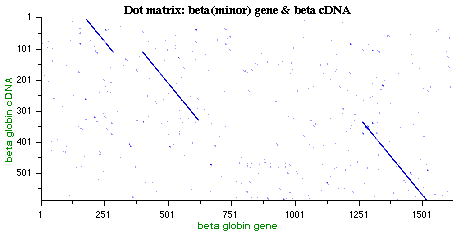
- Examples of DotPlot Visualization
Technique
- DotPlot @ Univ. Düsseldorf, Germany
- DotPlot: Dot matrix comparison of two sequences (not for Macintosh browser!)
Classic algorithms
- Needleman & Wunsch at EBI: Global alignment
- Smith & Waterman at EBI: Local alignment
Pairwise sequence comparisons
- BCM: Pairwise sequence comparisons: SIM, (ALIGN/LALIGN), BLAST2, LAP2, PGWISE, PCWISE
- ExPASy: Sequence comparisons: SIM, LALIGN (see below), Dotlet
- Sequence analysis Tools at EBI
- EMBOSS Pairwise Alignment Algorithms (global and local)
- Wise2 (basic) compares a protein sequence to a genomic DNA sequence, allowing for introns and frameshifting errors
- Wise2 (advanced)
- Wise2 Dna Block Aligner aligns two sequences under the assumption that the sequences share a number of colinear blocks of conservation separated by potentially large and varied lengths of DNA in the two sequences
- Wise2 PromoterWise compares two DNA sequences allowing for inversions and translocations, ideal for promoters
- ALIGN at Genestream
- BLAST 2 at NIH: Comparison of two sequence
- BLAST 2 at Genestream
- GLASS at MIT: GLobal Alignment SyStem
- LALIGN at EMBnet: Finds multiple matching subsegments in two sequences (SIM-based code)
- LALIGN at Genestream
- LFASTA at PBIL: Local Alignment Tool for Nucleic Sequences
- Palign (Stockholm): Protein alignment
- SIM at SIB/ExPASy: Alignment of two protein sequences with SIM, results can be viewed with LALNVIEW
- SIM4 at PBIL: Program to align cDNA and genomic DNA
- ToPlign (Toolbox for Protein ALignment) at Fraunhofer: Pairwise and multiple sequence comparison
- ToPlign: Login at BioSolveIT (OUTDATED)
- PRSS3 at EMBnet: evaluates the significance of a protein sequence alignment
Multiple sequence comparisons
- BCM: Multiple sequence comparisons: ClustalW, CAP, MAP, PIMA, MSA, BLOCK MAKER, MEME, Match-Box
- ExPASy: Sequence comparisons: ClustalW, KALIGN, MAFFT, Muscle, T-Coffee, MSA, DIALGN, Match-Box, Multalin, MUSCA (see below)
- ClustalW at EBI
- ClustalW at PBIL
- ClustalW at MyHits/SIB
- ClustalW at EMBnet
- COBALT at NIH: Multiple alignment incorporating pairwise constraints
- Coffee's at CNRS Marseille
- Coffee's at EBI
- Coffee's at SIB
- T-Coffee at BioAssist Wageningen
- DIALIGN at Bielefeld University: Multiple sequence alignment based on segment-to-segment comparison
- Kalign at EBI: A fast and accurate multiple sequence alignment algorithm
- Kalign at Karolinska
- MAFFT at EBI: Multiple Alignment using Fast Fourier Transform
- MAFFT at MyHits/SIB
- MAFFT at Kyushu University
- Match-Box at University of Namur (only proteins!)
- MSA at Genestream: Multiple Sequence Alignment
- Multalin at INRA
- Multalin at PBIL (only proteins!)
- MUSCA at IBM: Multiple sequence alignment using pattern discovery (only proteins!)
- MUSCLE at Drive5: MUltiple Sequence Comparison by Log-Expectation
- MUSCLE at EBI
- MUSCLE at BioAssist Wageningen
- PRALINE: PRofile ALIgNEment, at Vrije Universiteit Amsterdam
- PROBCONS: Probabilistic Consistency-based Multiple Alignment of Amino Acid Sequences, at Stanford University
- SAGA: Sequence Alignment by Genetic Algorithm (software for download)
- SATCHMO at Drive5: Simultaneous Alignment and Tree Construction using Hidden Markov mOdels (software for download)
- SSMAL at DKFZ: Shuffled Similarities with Multiple ALignments (software for download) (OUTDATED)
Profile comparisons
- COACH at Drive5: COmparison of Alignments by Constructing HMMs (software for download)
- COMPASS at University of Texas: COmparison of Multiple Protein Alignments with Assessment of Statistical Significance
- FFAS03: Fold and Function Assignment System
Genome comparisons:
- TaxPlot
- GRAPe at University of Oxford: Probabilistic whole-genome re-alignment (for download)
Similarity matrices:
- PAM250: Percent Accepted Mutation-Matrix (Dayhoff et al., 1978)
- BLOSUM62: Blocks Substitution-Matrix (Henikoff and Henikoff, 1992)
- More similarity matrices
- HELP to proper use of similarity matrices
Software for representation of a pairwise sequence alignment:
- LalnView at PBIL
- LalnView at SIB/ExPASy
Software for representation of a multiple sequence alignment:
- AMAS at EBI: Analyse Multiply Aligned Sequences (OUTDATED)
- AMAS at Dundee: Analyse Multiply Aligned Sequences
- Bork's alignment tools to enhance the results of multiple alignments (including consensus building)
- BoxShade 3.21 at EMBnet
- CINEMA 2.1 at Manchester: Color INteractive Editor for Multiple Alignments (incl. JAVA applet)
- CINEMA 5 / Utopia at Manchester
- CINEMA 5 / Utopia at Manchester (old website)
- ESPript at IBCP Lyon: Tool to print a multiple alignment
- JalView 1.3b at EBI (incl. download)
- JalView 1.8 at Dundee (discontinued, go to JalView 2)
- JalView (incl. recent downloads)
- Jevtrace2 at UC San Francisco
- MView at Pasteur
- STRAP: Interactive Structure based Sequences Alignment Program
- THoR: Thorough Homology Resource (OUTDATED)
Software to generate sequence logos:
- plogo: Protein sequence logos at CBS/Denmark
- slogo: RNA structure logos at CBS/Denmark
- GENIO/logo: Sequence logos
- WebLogo: Sequence logos at Berkeley
Short online course about sequence comparisons:
- Online course from "Biochemstry" (Jeremy M. Berg, John L. Tymoczko, Lubert Stryer; ISBN: 0-7167-3051-0)
Example of a pairwise sequence alignment:
- beta globin:
mvhltpeeks avtalwgkvn vdevggealg rllvvypwtq rffesfgdls tpdavmgnpk
vkahgkkvlg afsdglahld nlkgtfatls elhcdklhvd penfrllgnv lvcvlahhfg
keftppvqaa yqkvvagvan alahkyh
- Myoglobin:
mglsdgewql vlnvwgkvea dipghgqevl irlfkghpet lekfdkfkhl ksedemkase
dlkkhgatvl talggilkkk ghheaeikpl aqshatkhki pvkylefise ciiqvlqskh
pgdfgadaqg amnkalelfr kdmasnykel gfqg
- SIM homology search:
Gap open penalty: 24
Gap extension penalty: 4
Comparison Matrix: BLOSUM62
------------------------------------------------------------------------
23.8% identity in 122 residues overlap; Score: 91.0; Gap frequency: 0.0%
beta-Globin 25 GGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKG
Myoglobin 26 GQEVLIRLFKGHPETLEKFDKFKHLKSEDEMKASEDLKKHGATVLTALGGILKKKGHHEA
* * * ** * * * * * * * ** ** * *
beta-Globin 85 TFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAH
Myoglobin 86 EIKPLAQSHATKHKIPVKYLEFISECIIQVLQSKHPGDFGADAQGAMNKALELFRKDMAS
* * * ** * * * * *
beta-Globin 145 KY
Myoglobin 146 NY
*
------------------------------------------------------------------------
40.9% identity in 22 residues overlap; Score: 38.0; Gap frequency: 0.0%
beta-Globin 4 LTPEEKSAVTALWGKVNVDEVG
Myoglobin 3 LSDGEWQLVLNVWGKVEADIPG
* * * **** * *
------------------------------------------------------------------------
Gap open penalty: 12
Gap extension penalty: 4
Comparison Matrix: BLOSUM62
------------------------------------------------------------------------
25.5% identity in 145 residues overlap; Score: 103.0; Gap frequency: 1.4%
beta-Globin 4 LTPEEKSAVTALWGKVNVDEVGG--EALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKV
Myoglobin 3 LSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDKFKHLKSEDEMKASEDL
* * * **** * * * * ** * * * * * *
beta-Globin 62 KAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGK
Myoglobin 63 KKHGATVLTALGGILKKKGHHEAEIKPLAQSHATKHKIPVKYLEFISECIIQVLQSKHPG
* ** ** * * * * * **
beta-Globin 122 EFTPPVQAAYQKVVAGVANALAHKY
Myoglobin 123 DFGADAQGAMNKALELFRKDMASNY
* * * * * *
------------------------------------------------------------------------
Sequence examples:
- Database of amino acid sequences via Entrez
- Database of nucleotide sequences via Entrez
- Bacteriorhodopsin from Halobacterium salinarium: Seven-helix bundle protein
- TonB from Escherichia coli: Protein with N-terminal transmembrane helix
- Maltose-binding protein from Escherichia coli: Protein with N-terminal signal sequence for secretion into the periplasmic space
- OmpA from Escherichia coli: Two-domain protein: the N-terminal protein domain is embedded into the outer membrane in form of an 8-stranded β barrel while the C-terminal protein domain is found in the periplasmic space
Abbreviations:
- AMAS: Analyse Multiply Aligned Sequences
- BCM: Baylor College of Medicine
- BEAUTY: BLAST Enhanced Alignment Utility
- BLAST: Basic Local Alignment Search Tool
- BLOSUM: Blocks Substitution-Matrix
- CINEMA: Color INteractive Editor for Multiple Alignments
- COACH: COmparison of Alignments by Constructing HMMs
- COFFEE: Consistency based Objective Function For alignmEnt Evaluation
- COMPASS: COmparison of Multiple Protein Alignments with Assessment of Statistical Significance
- ExPASy: Expert Protein Analysis System
- FFAS03: Fold and Function Assignment System
- MAFFT: Multiple Alignment using Fast Fourier Transform
- MSA: Multiple Sequence Alignment
- NIH: National Institute of Health
- PAM: Percent Accepted Mutation
- PIMA: Pattern-Induced Multiple-sequence Alignment program
- PRALINE: PRofile ALIgNEment
- SAGA: Sequence Alignment by Genetic Algorithm
- SSMAL: Shuffled Similarities with Multiple ALignments
Latest update: October 14, 2009
Ralf Koebnik
Institut de recherche pour le dèveloppement
UMR 5096, CNRS-UP-IRD
911, Avenue Agropolis, BP 64501
34394 Montpellier, Cedex 5
FRANCE
Phone: +33 (0)4 67 41 62 28
Fax: +33 (0)4 67 41 61 81
Email: koebnik(at)gmx.de
Please replace (at) by @.
 Back to main page
Back to main page
|
